How to preprocess miRNA-seq with FastQC and cutadapt
Aim: conduct research on miRNA
Data: SRR5233942 .fastq directly from EBI
Please refer to SRA fastq Data Download
FastQC tool
To test fastq files’ quality.
Tips: 1.If you run FastQC with Linux command, run with file names; otherwise it will print errors
# Quality-check
/home/xuanzhan/Data/Downloads/FastQC/./fastqc SRR13338290.fastq
---- command output -----
SRR13338290_fastqc.html
SRR13338290_fastqc.zip
# Download .html file (find adaptor info) and open in chrome
scp cluster:/home/xuanzhan/Data/human/SRR13338290_fastqc.html ~/Desktop/
fastq_quality_filter -v -q 20 -p 80 -Q33 -i $id -o tmp ;
Cutadapt
1.Search for standard adapter sequence, Here is Illumina Adapter Sequences.
>"TruSeq Small RNA"
>1.RNA 5’ Adapter (RA5):
5’ GUUCAGAGUUCUACAGUCCGACGAUC
>2.**RNA 3’ Adapter (RA3)**:
5’ TGGAATTCTCGGGTGCCAAGG
>3.Stop Oligo (STP):
5’ GAAUUCCACCACGUUCCCGUGG
>4.RNA RT Primer (RTP):
5’ GCCTTGGCACCCGAGAATTCCA
>5.RNA PCR Primer (RP1):
5’ AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA
2.Run Cutadapt with -a/-g/-b parameter
“-j 0 “: to automatically detect the number of available cores, use -j 0
-a : 3’ adapter
-g : 5’ adapter
-b : not-sure 3’ or 5’, will try both
cutadapt -j 0 -a TGGAATTCTCGGGTGCCAAGG -o cut.fastq SRR5233942.fastq
Tips: If there is multiple adapters, user can provide a Fasta file with “-g file:barcodes.fasta” ,but doesn’t work, when I test files; refer to Cutadapt:multipe-adapters.
# cut
cutadapt -j 0 -g AGGCAGTGTAGTTAGCTGATTGCATCTCGTATGCCGTCTT -g TAGCTTATCAGACTGATGTTGAATCTCGTATGCCGTCTTC -M 25 -o cut.fastq SRR13338290.fastq
# FastQC
/home/xuanzhan/Data/Downloads/FastQC/./fastqc -t 6 cut.fastq
# Download FastQC results (local Terminal)
scp cluster:/home/xuanzhan/Data/human/cut_fastqc.html ~/Desktop/
3.After adapter cut, all reads are received. so need to Fliter:
-m LENGTH: Discard processed reads that are shorter than LENGTH.
-M LENGTH: Discard processed reads that are larger than LENGTH.
–untrimmed-output FILE: Write all reads without adapters to FILE (in FASTA/FASTQ format) instead of writing them to the regular output file.
如何过滤长度?
###cutadapt shell script
#install on sever node (vulcan3)
#通过 pip3 install --user cutadapt 安装在目录 ~/.local/lib/python3.6/site-packages
cutadapt -a TGGAATTCTCGGGTGCCAAGG --untrimmed-output untrimmed.out -o out.fastq SRR5233942.fastq
cutadapt -a TATGGAGTTCTACAGTCCGACGATCTA --untrimmed-output untrimmed.out -o out-2.fastq out.fastq
Trimmomatic (java tools)
java -jar /home/xuanzhan/Data/Downloads/Trimmomatic-0.39/trimmomatic-0.39.jar SE -phred33 /home/xuanzhan/Data/miRNA/salmon/SRR866579.fastq /home/xuanzhan/Data/miRNA/salmon/cut_579.fastq ILLUMINACLIP:/home/xuanzhan/Data/Downloads/Trimmomatic-0.39/adapters/RNA3-adapter.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:18
# check by fastqc
/home/xuanzhan/Data/Downloads/FastQC/./fastqc cut_573.fastq
参数说明
Phred33 设置碱基的质量格式,默认的是-phred64。
ILLUMINACLIP:$adapter.fa:2:30:10 adapter.fa为接头文件,2表示最大mismatch数,30表示palindrome模式下碱基的匹配阈值,10表示simple模式下碱基的匹配阈值。
LEADING: 3 表示切除reads 5’端碱基质量低于3的碱基。
TRAILING:3 表示切除3’ 端碱基质量低于3的碱基。
SLIDINGWINDOW:4:15 表示以4个碱基为窗口进行滑动,切除窗口内碱基平均质量小于15的。
MINLEN:36 丢弃以上步骤处理后,序列长度小于36的reads。
Another tool
fastx_trimmer -v -f 1 -l 27 -i tmp -Q33 -z -o
去接头工具比较
cutadapt优点(推荐使用)
- 参数简洁
- 接头去的很干净
Trimmomatic
- 参数比较繁琐
- 同样接头文件,在默认参数的情况下无法去除彻底
Case Study
A preprocess script for a raw miRNA seq (SRR5233942)
fastqc SRR5233942.fastq
# 从Adapter Content 和 overrepresented sequences 中找到接头序列
# cutadapt 切除接头 和 未被处理序列
cutadapt -a TGGAATTCTCGGGTGCCAAGG --untrimmed-output untrimmed.out -o out.fastq SRR5233942.fastq
cutadapt -a TATGGAGTTCTACAGTCCGACGATCTA --untrimmed-output untrimmed.out -o out-2.fastq out.fastq
# 按miRNA数据库统计的长度范围取16-35长的seq
cutadapt -m 16 -M 35 -o output.fastq input.fastq
对整理后的数据进行不同长度k-mer频率统计: 总reads数:18154(18-25bp)
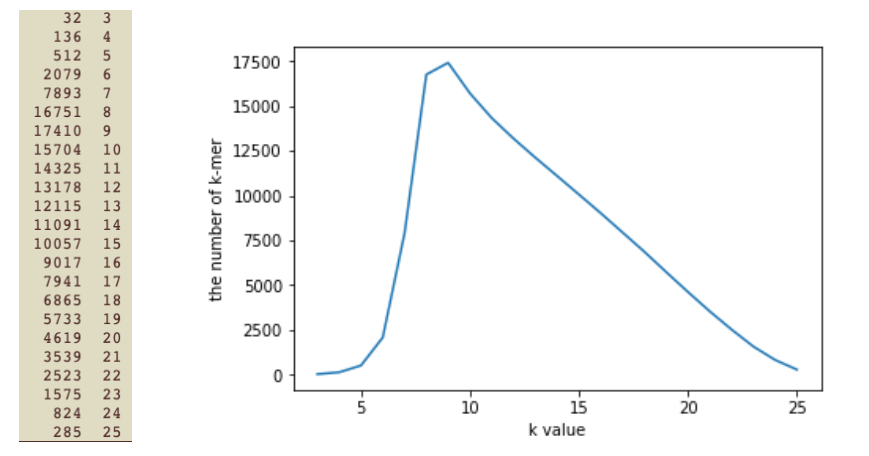
import matplotlib.pyplot as plt
count = []
k = []
with open('tmp.out','r') as f:
for line in f:
count.append(int(line.split( )[0]))
k.append(int(line.split( )[1]))
plt.plot(k,count)
plt.xlabel('k value')
plt.ylabel('the number of k-mer')
plt.show()
A script for download miRNA-seq of Salmon
#!/bin/bash
id=(573 579 583 587 589 590 605 611 612 613 614 615)
for i in $(seq 0 11);
do
echo "SRR${id[i]} is precessing";
/home/xuanzhan/Data/bin/./fastq-dump SRR866${id[i]}
/home/xuanzhan/Data/Downloads/FastQC/./fastqc SRR866${id[i]}.fastq
cutadapt -a TGGAATTCTCGGGTGCCAAGG --untrimmed-output untrimmed.out -o out.fastq SRR866${id[i]}.fastq
cutadapt -m 18 -M 25 -o cut_${id[i]}.fastq out.fastq
/home/xuanzhan/Data/Downloads/FastQC/./fastqc cut_${id[i]}.fastq
done
rm *.zip
rm out.fastq
echo "Download finish";
Reference
Support or Contact
xzhangxmu@gmail.com