使用KMC3工具统计k-mer频率
KMC3工作原理
Two-stage processing scheme:
1.split reads into several hundred bins(disk files), according to super k-mers
2.Bins are sorted one by one to remove duplicates
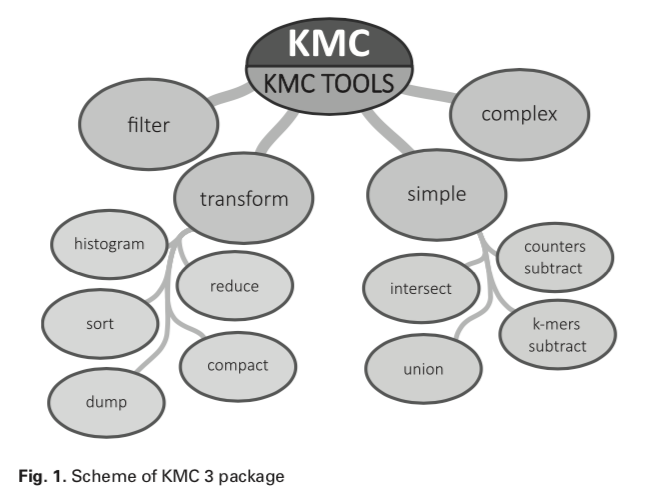
containing various tools:
1.Filter :extract from fastq files the reads satisfying some criteria.
2.transform : modify KMC database, remove too frequency/rare k-mers, remove counters?, build histogram??
3.Intersect
4.Union
Evaluation with another three tools:
- DIAMUND (2014)
- NIKS (2013)
- GenomeTester4 (2015)
KMC3使用及参数说明
kmc : 数k-mer,输出为两个压缩文件(后缀分别为:kmc_pre; kmc_suf)
kmc_dump : 解压输出k-mer及频率信息
参数说明
-ci(value) : exclude k-mers occuring less than (default is 2)
-cx(value) : exclude k-mers occuring more than
for i in $(seq 6 7);
do
./kmc -k${i} -ci1 ~/Data/miRNA/18-25.fastq k${i} ./
./kmc_dump k${i} kc${i}
#按频率排序,删除中间文件,只保存排序后文件
sort -nk2 -r kc${i} > ${i}.freq
rm k${i}*
rm kc${i}
echo "${i}-mer counting finishing /n"
done
具体问题
1.对于一个k-mer ,是否考虑互补序列? 频率如何算?
假设k-mer (AGGTT), 他的逆序互补序列为(AACCT)
k-mer counters usually do not distinguish between direct k-mers and their reverse complements and collect statistics for canonical k-mers. The canonical k-mer is lexicographically smaller of the pair: the k-mer and its reverse complement.
只取(字母序较小的那条)作为代表。(均被统计,且分别记录各自出现频率;)
-
k为偶数时:本链与反向互补链可能相同
-
k为奇数时:本链与反向互补链不同
2.测序数据是否需要区别本链和反向互补链? 有需要区别链么?
- 测序中的正负链 (Forward & reverse)
测序之前,要先将序列打断,进行建库。在建库的时候是不区分正负链的,所以在后续测序过程中产生序列也不包含正负链信息。
当测的read要往参考基因组上比对时,参考基因组中每条染色体只有一条序列,这条序列即为正链序列,常用‘+’表示,与正链互补的序列为负链,用‘-’表示。
对一条染色体而言,只有一条正链序列,互补的即为负链。
文献或者数据库中提到的基因所在strand,即为这里所提的正负链。
- 转录翻译中的正负链(正义链 sensestrand & 反义链 anti-sensestrand)
根据中心法则,DNA转录成mRNA,mRNA翻译成蛋白质。mRNA只能从一条DNA链,根据碱基互补配对转录形成一条RNA链,这条辅助RNA转录的DNA链叫anti-sense strand无义链或者叫template strand模板链.该模版链与mRNA序列互补。
另一条与mRNA链序列相同的DNA链(除了T被U取代),被定义为正链(非模版链),即RNA序列相同的那一个dna单链。
相对应的链就是负链(模版链)。
- 答:测序数据本身没有正负链标记,在进行map后才会被区别;仅测序后的fastq文件是没有正负链差别的。
3.miRNA测序怎么做的?
4.miRNA去接头后的数据可以直接在miRNA数据库中查找到么?
使用BFCounter工具计算k-mer频率
- ./BFCounter count
- ./BFCounter dump
for i in $(seq 6 25);
do
/home/xuanzhan/Data/Downloads/BFCounter/./BFCounter count -k ${i} -n 999999999 -o /home/xuanzhan/Data/miRNA/exp/BFC_${i} /home/xuanzhan/Data/miRNA/exp/18-25.fastq
/home/xuanzhan/Data/Downloads/BFCounter/./BFCounter dump -k ${i} -i /home/xuanzhan/Data/miRNA/exp/BFC_${i} -o /home/xuanzhan/Data/miRNA/exp/BFC_dump_${i}
sort -nk2 -r BFC_dump_${i} > BFCsorted_${i}
done
rm BFC_*
Related work
#bfc,coral运行成功;bfc未纠错(可能应该尝试其他参数)
cd /home/xuanzhan/Data/bin
./bfc -t28 18-25.fastq > bfc.fq
./coral -fq 18-25.fastq -o coral.fq
#karect没成功
karect -correct -inputfile=/dev/shm/p_t_chr1.fq -celltype=diploid -matchtype=hamming -resultprefix=karect_
mv /dev/shm/karect_p_t_chr1.fq /dev/shm/karect_chr1.fq
#bcool 没成功
mkdir bcool
/home/xuanzhan/Data/Downloads/BCOOL/./Bcool -u ${id[${i}]}.fa -o bcool -t 28
mv bcool/reads_corrected.fa L_bcool_${id[${i}]}.fa
rm -rf bcool/*
Reference
| [链特异RNA-seq数据不这么看就浪费了 | antisense上的lncRNA-seq](http://blog.sciencenet.cn/home.php?mod=space&uid=3372875&do=blog&id=1090305) |
| [biostar handbook(五) | 序列从何而来和质量控制](https://www.jianshu.com/p/ec4d63a8fa9f) |